Structural bioinformatics: proteins in life and disease
We are interested in developing the sequnce/structure computational models for prediction of protein interactions, including protein-protein, protein-DNA and small molecule-protein interactions. Our special interest is in reconstruction of the interaction networks and understanding the perturbations of molecular interactions in disease conditions.
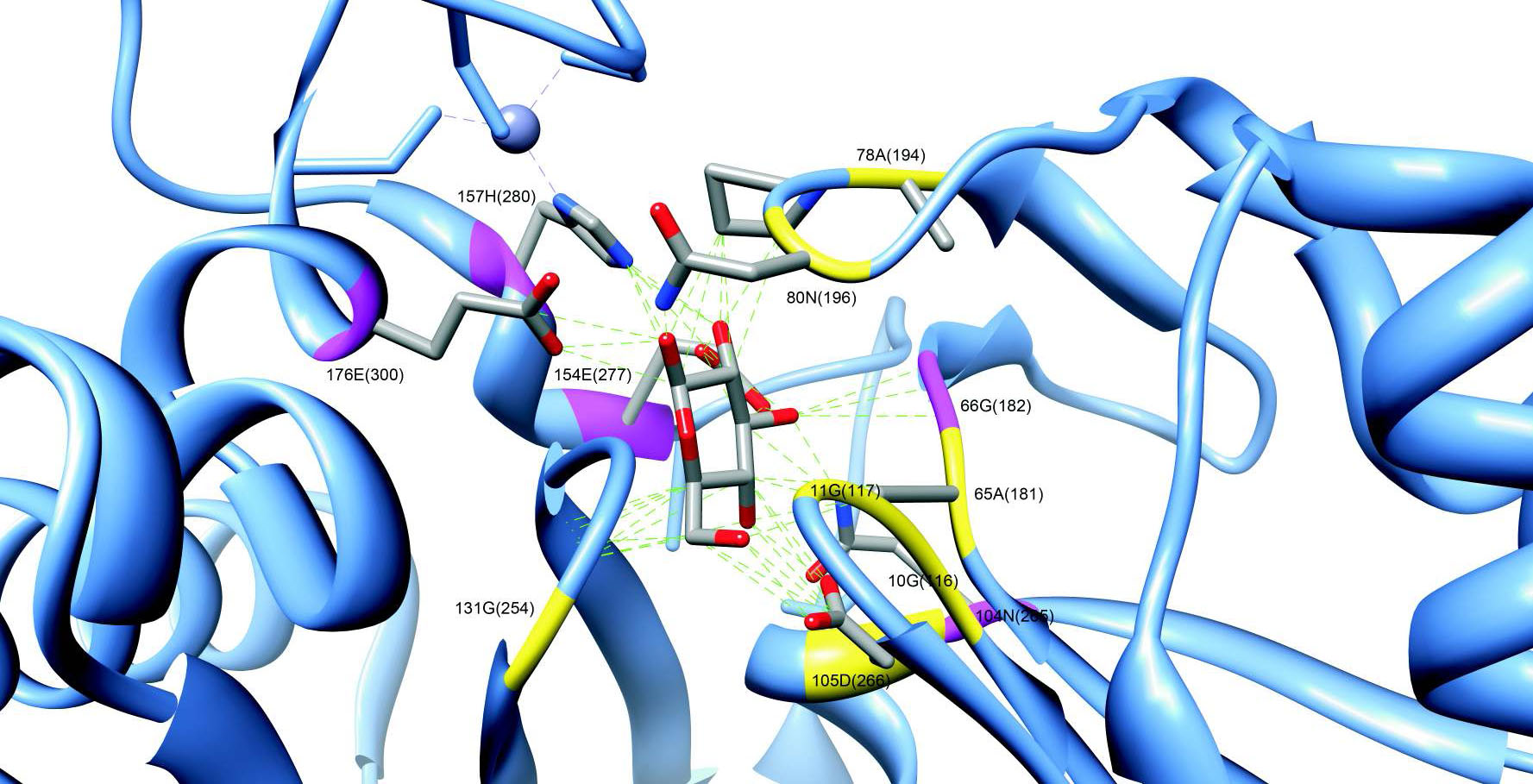
Prediction of protease substrates
Proteases are enzymes catalyzing a cleavage of the polypeptide chain. In human more than 500 proteases are known. They participate in such processes as blood coagulation, immune response and digestion. It is known that in many disease the conventional proteolytic pathways are disrupted. For example, in cancer abnormal protease activity promote metastatic progression. We are interested in the prediction of protease substrates for the elucidation of the networks of proteolytic interaction.
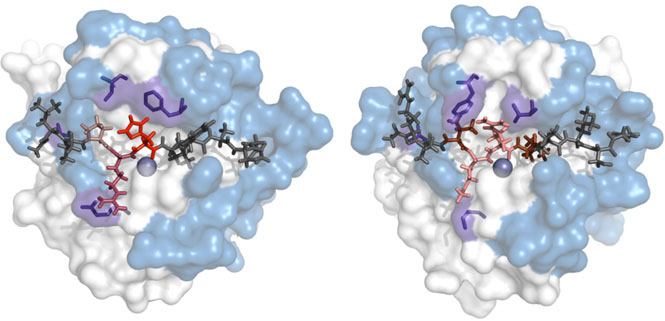
Structural basis of protein specificity
One of the important questions in molecular biology is the mechanisms of the recognition of small molecules by proteins. Our approach for studying such mechanisms is the comparison of the orthologous proteins having different specificity. Such analysis could identify the sequence and structural specificity determinants representing the knowledge that can be further used for developing of the computational models for protein specificity prediction.
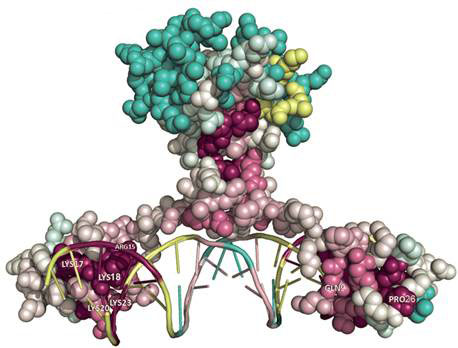
Regulation of gene expression by transcription factors
We are interested in computational prediction of interaction of transcription factors, the proteins that orchestrate the gene expression, with DNA. Transcription factors interact with the short portion of DNA called regulatory sites, which usually represent a small variation of the particular nucleotide sequence. We are interested in developing the sequence/structure models for prediction of regulatory sites and its application for the reconstruction of regulatory networks.
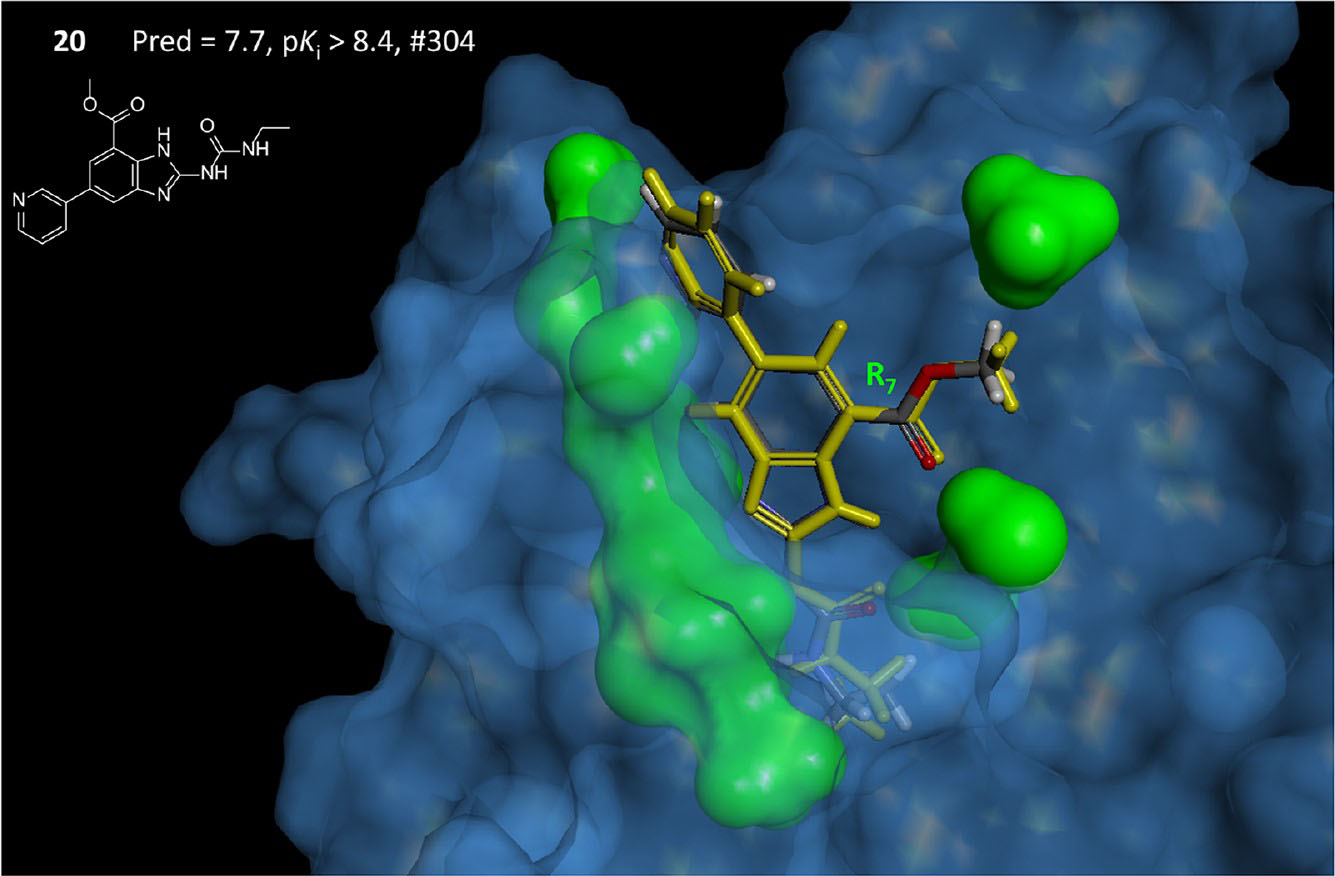
Computational methods for drug discovery
The usual initial step in a complex multistep process of the drug development is the screening of the large molecular libraries of chemical compounds against the target protein for the identification of drug candidates. As the size of modern compound libraries are large enough the screening becomes expansive and slow procedure. The predictive quantity-structure relation models (QSAR) intended to make this search more rational, saving time and resources. We are interested in developing and application of 2D and 3D QSAR computational models for the drug development.
Cancer bioinformatics
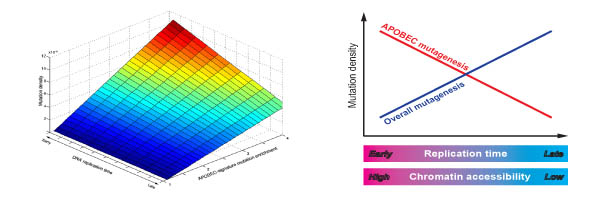
Cancer genome analysis recently become available with an advances in the sequencing technologies. Computational analysis of mutations in cancer genomes including their associating with other known genome features could shed the light on mechanisms of the cancer mutagenesis. Our latest collaborative project devoted to the analysis of the mutations induced by the human immune system proteins from the APOBEC family revealed its association with the several epigenomic features.
Biomedical imaging
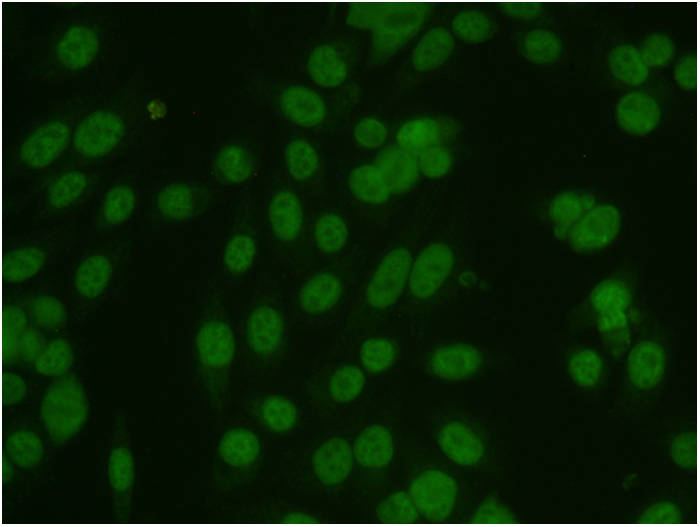
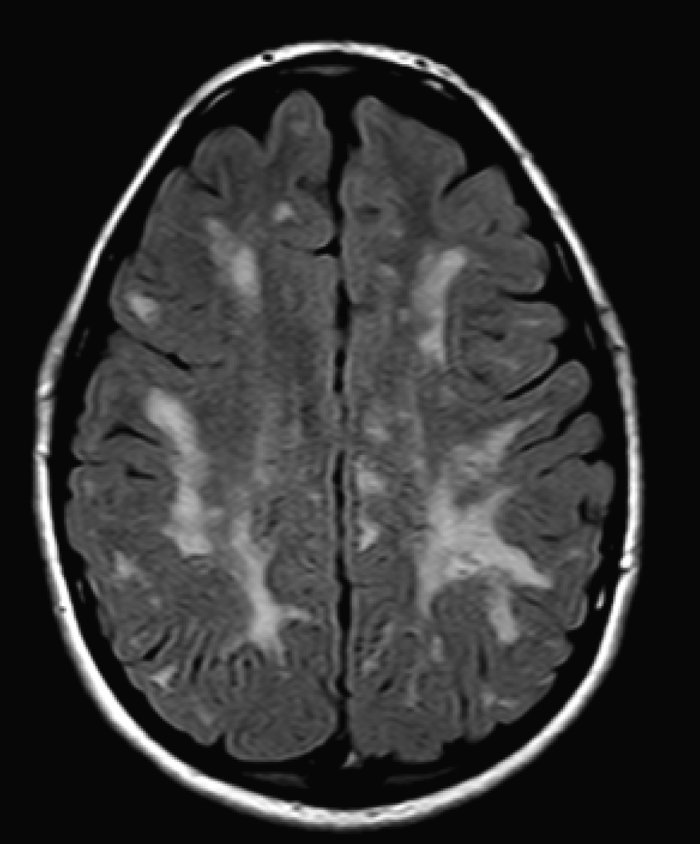
Biomedical image analysis as rule aimed to extract from the acquired biological or medical image the object of interest for recognizing desired properties of this object. The process of the detection of the object's contour in image is calling the image segmentation. We are interested in the developing methods for the segmentation of various biomedical, both color and grayscale, images. Our recent projects in this field include the project of image classification for the ANA HEp-2 medical test, which recognize type of the human autoimmune disease, and the project of mapping of anatomical structures in brain images obtained by various neuroimage techniques.