Структурная биоинформатика: функции белков в норме и при патологии
Областью наших интересов является разработка вычислительных моделей предсказания белковых взаимодействий, включая взаимодействия белок-белок, белок-ДНК и белок-малые молекулы. Нас также интересует реконструкция сетей белковых взаимодействий и нарушения в их структуре в условиях болезни.
Предсказание субстратов протеаз
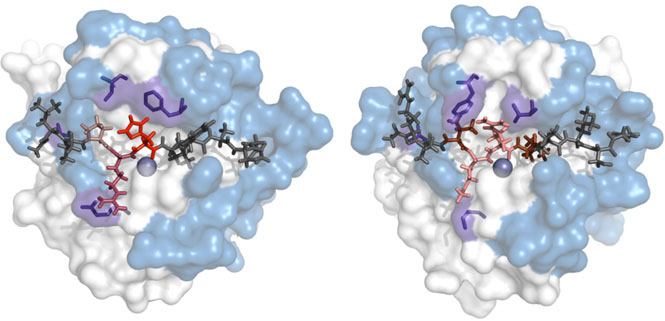
Протеазами называются ферменты катализирующие расщепление пептидной связи. В геноме человека известно более 500 генов, кодирующих протеолитические ферменты. Протеазы участвуют в таких процессах, как коагуляция крови, пищеварение и иммунный ответ. Известно, что при многих заболеваниях в организме человека происходят нарушения в протеолитических каскадах. Так, например, при раке патологическая активность протеаз может приводить к метастазированию опухолей. Областью наших интересов является предсказание субстратов протеаз вычислительными методами для последующей экспериментальной проверки и составления подробных карт протеолитических взаимодействий в организме человека.
Структурные основы специфичности белков
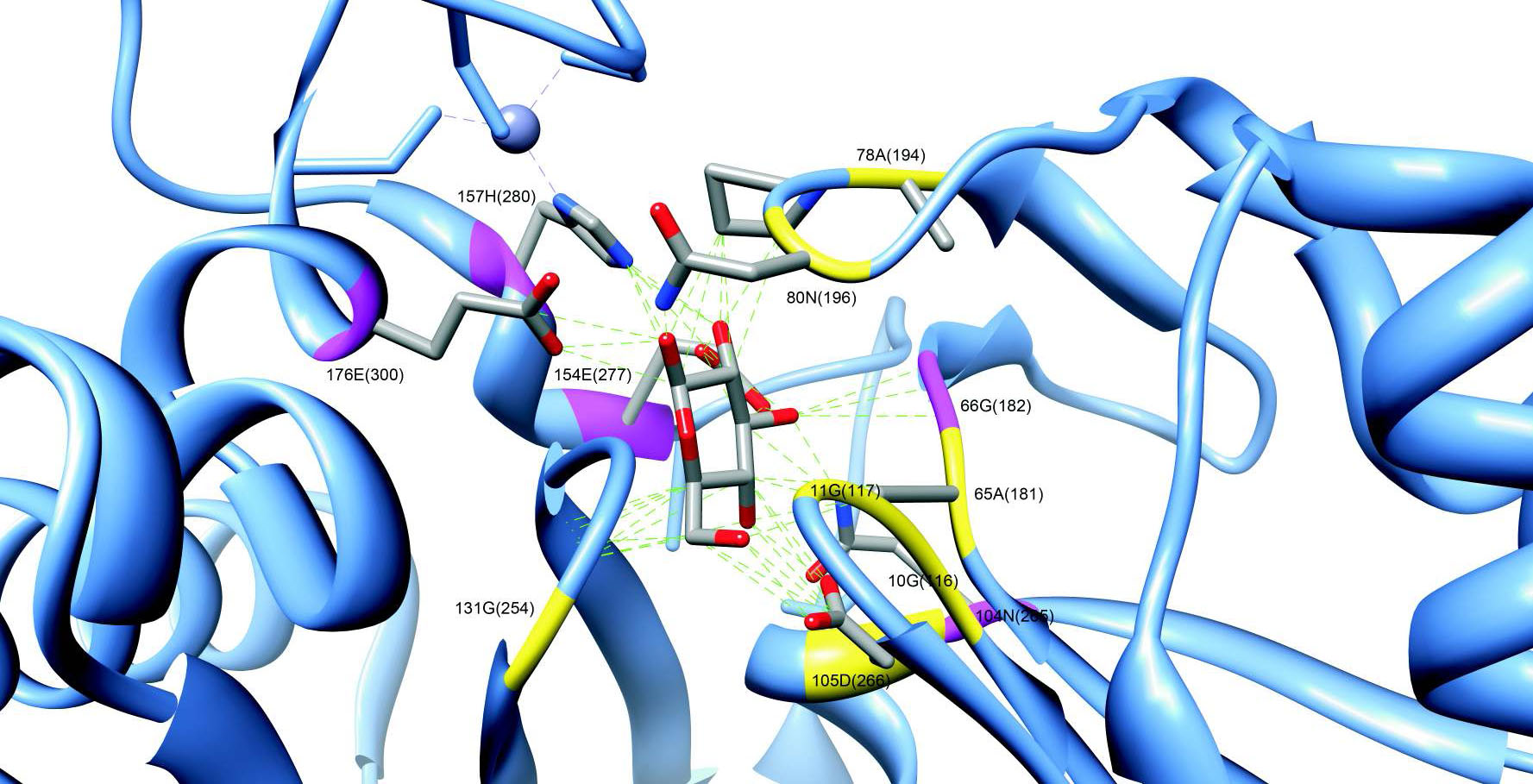
Одним из важных вопросов в молекулярной биологии является изучение механизмов распознавания белками малых молекул. Наш подход для изучения данных механизмов заключается в сравнении близкородственных белков, имеющих различную специфичность. Такой анализ позволяет определять структурные детерминанты, ответственные за специфичность белков, и далее применять полученные знания для разработки вычислительных моделей предсказания специфичности белков.
Регуляция экспрессии генов факторами транскрипции
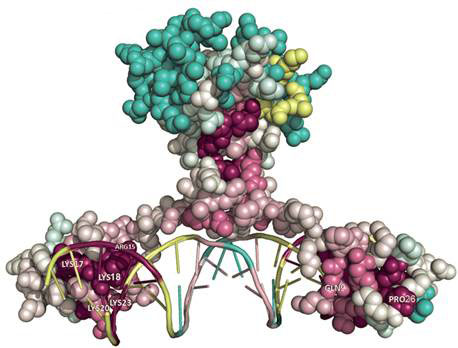
Нас интересует предсказание вычислительными методами взаимодействия транскрипционных факторов - белков, управляющих экспрессией генов - с ДНК. Транскрипционные факторы как правило взаимодействуют с небольшими участками молекулы ДНК называемыми регуляторными сайтами, которые для одного транскрипционного фактора представляют собой небольшие вариации определенной последовательности ДНК. В область наших интересов входит разработка моделей предсказания регуляторных сайтов и их приложение для реконструкции регуляторных сетей.
Вычислительные модели для поиска новых лекарств
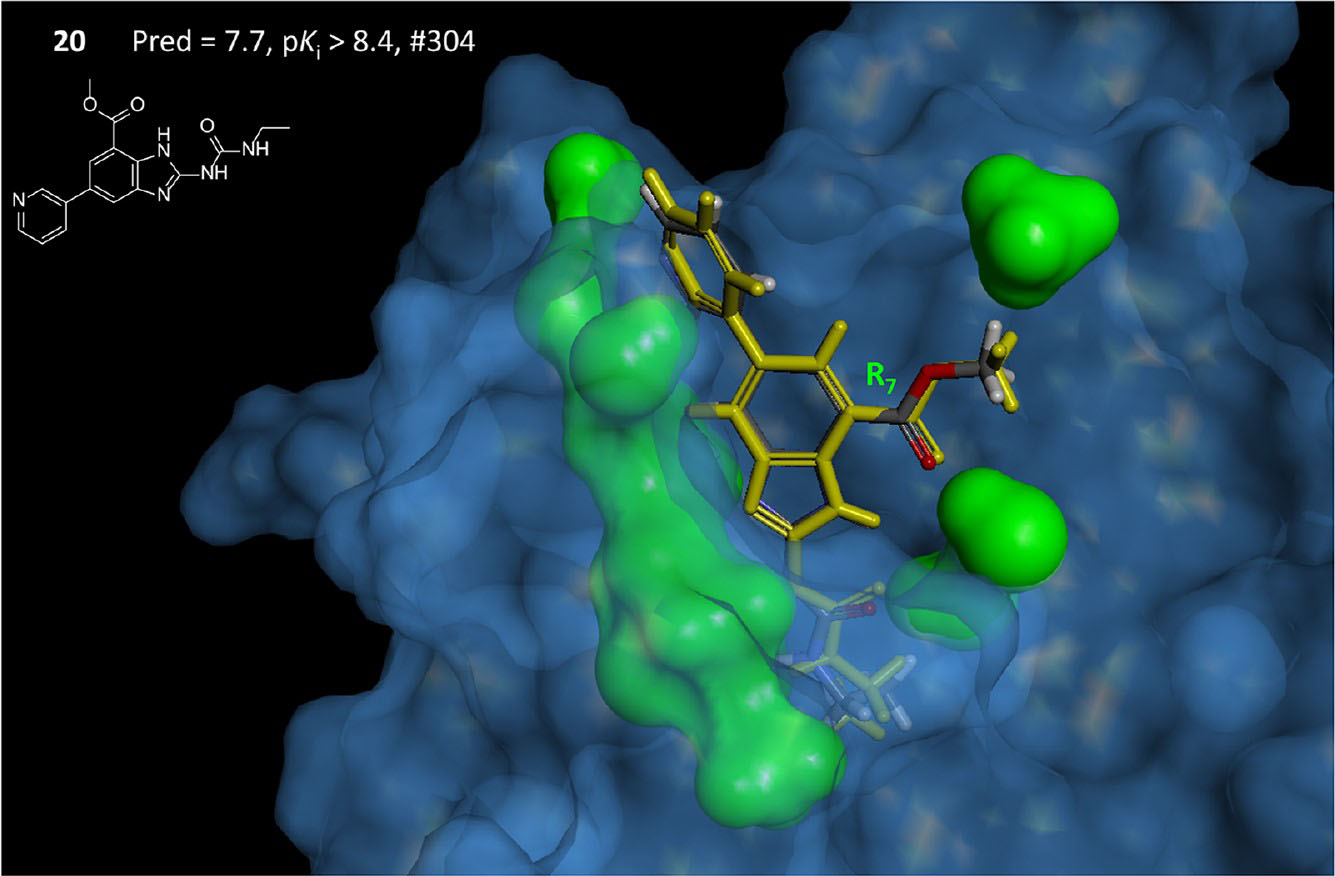
Первоначальным шагом в многоступенчатом процессе разработки лекарственных препаратов, как правило, является скрининг библиотек малых молекул относительно ингибируемого белка для определения молекул-кандидатов для дальнейшей оптимизации. Так как текущие размеры библиотек малых молекул довольно велики, то последовательный скрининг библиотек становится ресурсозатратной и медленной процедурой. Предсказательные модели количественных соотношений структура-активность (QSAR) могут сделать данный поиск более рациональным, а следовательно более быстрым и менее трудоемким. В область интересов нашей группы входит создание 2D и 3D QSAR моделей, применяемых при разработке лекарственных препаратов.
Биоинформатика в онкологии
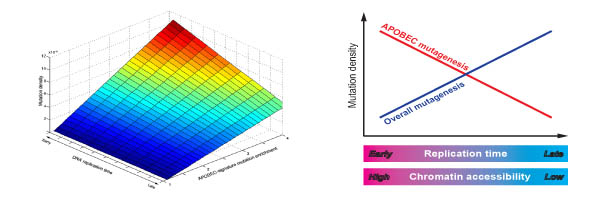
В связи с значительным прогрессом в технологиях секвенирования в последнее время стало возможным получение полных геномов клеток злокачественных опухолей. Вычислительный анализ мутаций в данных геномах, включая поиск ассоциаций мутаций с другими свойствами генома в позициях мутаций, может способствовать лучшему пониманию мутагенеза клеток злокачественных опухолей. В рамках проекта посвященного анализу мутаций, индуцируемых белками иммуной системы человека из семейства APOBEC, нами, совместно с коллабораторами, была обнаружена корреляция позиций мутаций с определенными эпигеномными свойствами.
Анализ биомедицинских изображений
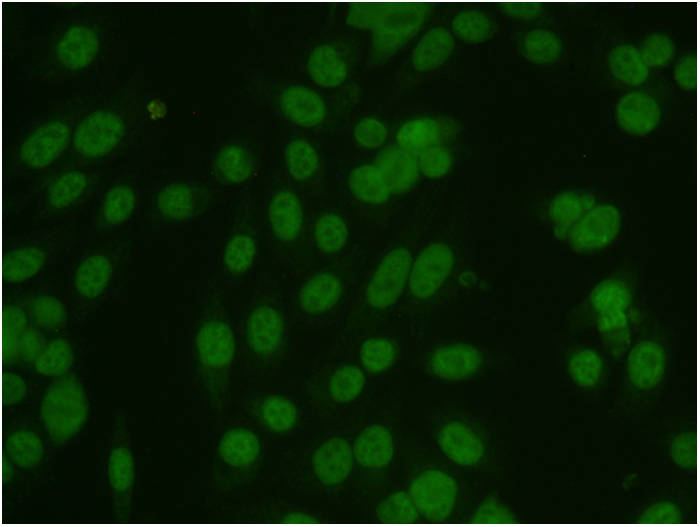
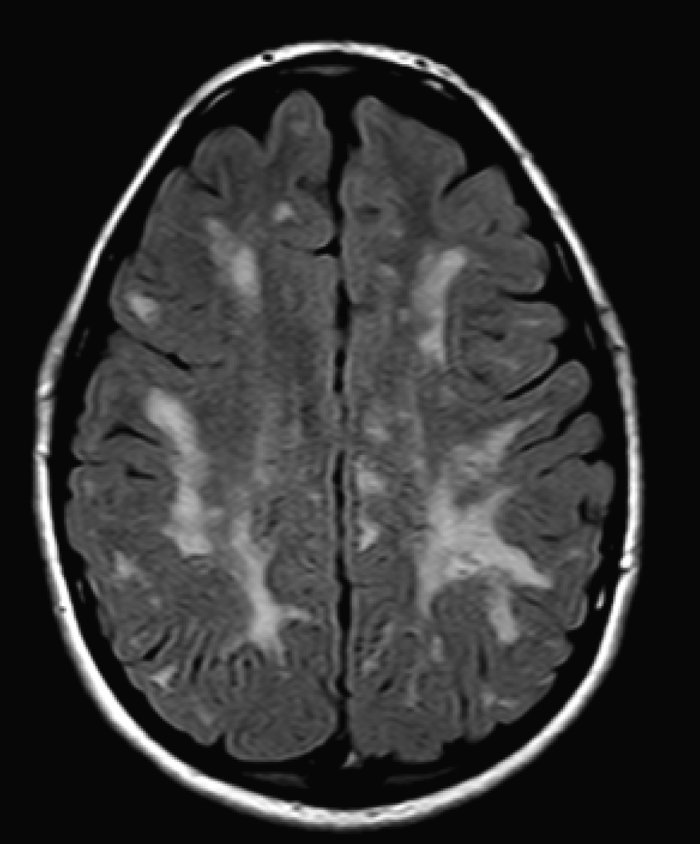
Методы анализа биомедицинских изображений, как правило, предназначены для обнаружения на изображении интересуемого объекта и определения его свойств. Процесс определения границ объекта называют сегментацией изображения. Область наших интересов включает сегментацию цветных и полутоновых биомедицинских изображений. Наши последние проекты в данной области включают метод классификации изображений медицинского теста ANA HEp-2 и проект автоматического картирования анатомических структур головного мозга на основе данных нейровизуализации.